
Top Essay Writers
Our top essay writers are handpicked for their degree qualification, talent and freelance know-how. Each one brings deep expertise in their chosen subjects and a solid track record in academic writing.
Simply fill out the order form with your paper’s instructions in a few easy steps. This quick process ensures you’ll be matched with an expert writer who
Can meet your papers' specific grading rubric needs. Find the best write my essay assistance for your assignments- Affordable, plagiarism-free, and on time!
Posted: February 14th, 2022
Amyotrophic lateral sclerosis or commonly known as ALS is one of the major neurodegenerative diseases alongside Alzheimer’s disease and Parkinson’s disease in the United States. A-myo-trophic is derived from the Greek language. A meaning no, Myo meaning muscle, and Trophic meaning nourishment that translates to No-muscle-nourishment. With no nourishment, the tissues degenerate leading to scarring or sclerosis of the region. Lateral indicates the location in the spinal cord, responsible for effective functioning of neurons. [1] ALS is a progressive disorder that involves degeneration of the upper motor neurons (UMN) in the frontal lobe of the brain and the lower motor neurons (LMN) in the brain stem and the spinal cord.
In ALS, as motor neurons die, a person loses the ability to walk, speak, swallow, and breathe. As the degeneration advances, the muscles gradually weaken and atrophies, losing its ability to control voluntary movements. ALS may rarely impair a person’s mind or personality, but people with ALS develop cognitive problems involving memory, speech fluency, and decision-making. ALS is usually fatal within 2-5 years of diagnosis. [2]
Students often ask, “Can you write my essay in APA or MLA?”—and the answer’s a big yes! Our writers are experts in every style imaginable: APA, MLA, Chicago, Harvard, you name it. Just tell us what you need, and we’ll deliver a perfectly formatted paper that matches your requirements, hassle-free.
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neuromuscular disease. Jean-Martin Charcot first described the disease in 1869. He used ALS as a prototypic example of his research techniques to coin the term “méthode anatomoclinique.” This method provided a disciplined and systematic approach to classify neurological diseases based on integrating clinical signs and anatomical lesions. [3] Because of Charcot’s fundamental contributions, the term “Charcot’s disease” is used as a synonym for amyotrophic lateral sclerosis.
ALS is also referred to as ‘Lou Gehrig’s Disease’ as it caused the death of the New York Yankees baseball player, Lou Gehrig in 1939. ALS can affect any human being regardless of their age, sex, or ethnic groups. For example, the famous astrophysicist Stephen Hawking was diagnosed with ALS at a young age and has survived for over 50 years. Mao Zedong, founder of the People’s Republic of China, Lane Smith- an American actor, O.J. Brigance- a professional football player, have been victims of ALS. [1]
In the summer of 2014, social media was taken by storm with videos of people pouring ice water on themselves for the Ice Bucket Challenge. This initiative was introduced by Pete’s Frates, a professional baseball player, who helped in increasing awareness for ALS and raised millions of dollars for research. The Ice Bucket Challenge was an enormously successful Internet phenomenon accepted by many actors, philanthropist, artists, and athletes. [4] Over 17 million people uploaded their challenge videos to various social media sites, and these videos were watched by 440 million people! The ALS Association collected $115 million in a six-week span from the ALS Ice Bucket Challenge. The ALSA reports that a majority of this fund was allocated for research and the rest was used for patient and community services, education, and fundraising. [4], [5]
ALS is the most frequent adult-onset motor neuron disease. It is characterized by both upper and lower motor neuron degeneration and has a median survival of 2–5 years. [2] The worldwide annual incidence of ALS is about 1.9 per 100,000 individuals. Since, almost all patients with ALS die of their disease, mortality rates for ALS individuals remains constant. As per the recent analysis, the number of ALS cases worldwide is predicted to rise by 69% over the next 25 years. According to the United Nations, the number of individuals above age 60 is expected to increase rapidly. This increase is particularly due to improving healthcare and economic conditions among developing nations. [6]
ALS can strike at any age, although symptoms develop as one grows older. Mean age at onset is 58 to 63 years for sporadic ALS and 40 to 60 years for familial ALS. [2] As per the Centre for Disease Control and Prevention results, individuals with those aged 18 to 39 years had the least prevalence rate (0.5 per 100,000 persons), and the age group 70 to 79 years had the highest prevalence rate (17.0 per 100,000 persons). [7], [8] The prevalence of ALS increases with age.
It is observed that men are at a higher risk to develop ALS than women, leading to a male-to-female ratio of 1.2–1.5. [7] Although as age increases, the incidence of ALS between men and women disappears. Few studies suggest that military veterans are twice more prone to ALS, especially those deployed during the Gulf War. [2] Possible risk factors for veterans include exposure to lead, pesticides, and diverse environmental toxins.
Our pricing starts at $10 per page for undergrad work, $16 for bachelor-level, and $21 for advanced stuff. Urgency and extras like top writers or plagiarism reports tweak the cost—deadlines range from 14 days to 3 hours. Order early for the best rates, and enjoy discounts on big orders: 5% off over $500, 10% over $1,000!
ALS can affect any person with no racial, ethnic, or socioeconomic boundaries. The prevalence rate for Caucasians was 2-fold greater than in African-Americans. Caucasians have a prevalence rate of 4.2 per 100,000 as compared to 2.0 per 100,000 for African-Americans. [7] Some geographic regions have an unusually high incidence of ALS, specifically Guam and the Kii Peninsula in Japan. The incidence of ALS in these regions is high due to environmental factors, especially a neurotoxic non-protein amino acid, β–methylamino-L-alanine (BMAA) produced in the seeds of Cycas micronesica. [2] It is hypothesized that patients in these regions may have a genetic susceptibility because of their inability to prevent BMAA accumulation.
Amyotrophic lateral sclerosis is a condition involving both UMN and LMN. The progression and spread of the disease can be both local and between neuro-anatomically linked regions. The identification of specific phenotypes helps in prognosis and survival, and also for their enrolment in clinical trials. The clinical manifestation of ALS helps in learning about the progression of the disease in an affected individual. The important presentations of ALS are described below;
Limb-onset ALS is the dominant feature with 70% of the cases among patients. [9] The main clinical feature in Limb-onset ALS is a combination of UMN and LMN damage involving brainstem and spinal cord regions. Patients with lower limb onset may complain of tripping, stumbling while walking or running. Patients with upper limb onset face difficulty in performing actions such as eating, writing, or picking up small objects.
Patients with bulbar onset present both upper and lower motor neuron signs. Dysarthria is a characteristic feature in patients with bulbar-onset ALS. Bulbar upper motor neuron symptoms include speech problems such as slurring, hoarseness, drooling, and distorted speech are typical symptoms. [10] Bulbar lower motor neuron signs include tongue wasting, fasciculations, and flaccid dysarthria. Some ALS patients show pseudo-bulbar symptoms like exaggerated involuntary emotional responses. Episodes of intense laughter may be followed at once by tears.
Yes, totally! We lock down your info with top-notch encryption—your school, friends, no one will know. Every paper’s custom-made to blend with your style, and we check it for originality, so it’s all yours, all discreet.
Primary lateral sclerosis is a variant of ALS with UMN involvement. It is a slow progressive type but affects the whole body. It is a rare motor neuron disease and spreads from the bulbar region to limbs. [9], [10] Primary lateral sclerosis is characterized by spasticity, weakness, pathologically hyperreflexia, and pseudo-bulbar speech.
Progressive muscular atrophy is another variant of ALS, represented by progressive LMN signs without clinical evidence of UMN dysfunction. [9] Symptoms of this disease are fasciculation, atrophy, and muscle weakness.
Although the etiology of ALS is not entirely understood, it can be classified into Familial ALS and Sporadic ALS. This section explains in brief the genetic and non-genetic causative agents for ALS.
No way—our papers are 100% human-crafted. Our writers are real pros with degrees, bringing creativity and expertise AI can’t match. Every piece is original, checked for plagiarism, and tailored to your needs by a skilled human, not a machine.
5.1 Familial ALS (FALS)
Familial ALS means that there is more than one occurrence of the disease in a family. FALS represents about 5 ~ 10% of all ALS cases diagnosed. [11] FALS can be further categorized by mode of inheritance and sub-categorized by the specific gene. Over 25 causative genes have been linked to hereditary ALS. FALS can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner depending on the gene involved.
5.1.1 Autosomal Dominant (AD)
Autosomal dominance is a characteristic pattern of inheritance wherein a single copy of the disease-associated gene is sufficient to cause the disease. A few examples of AD genes are Superoxide dismutase1 (SOD1), Fused in sarcoma (FUS), TAR DNA-binding protein (TARDBP), and Chromosome 9 open reading frame 72 (C9orF72).
We’re the best because our writers are degree-holding experts—Bachelor’s to Ph.D.—who nail any topic. We obsess over quality, using tools to ensure perfection, and offer free revisions to guarantee you’re thrilled with the result, even on tight deadlines.
5.1.2 Autosomal Recessive (AR)
In an autosomal recessive inheritance, the disease-associated gene must be inherited from both parents in order for an individual to develop the condition. ALSIN and OPTN (Optineurin) genes are common examples of AR inheritance in ALS.
5.1.3 X-Linked
In X-linked dominant inheritance, the gene responsible for the condition is located on the X chromosome. Mutation in either male and female copy of the gene can cause the disorder. Pathogenic variants in gene UBQLN2 (Ubiquilin 2) is related to X-linked dominant ALS.
Our writers are top-tier—university grads, many with Master’s degrees, who’ve passed tough tests to join us. They’re ready for any essay, working with you to hit your deadlines and grading standards with ease and professionalism.
5.2 Sporadic ALS (SALS)
Sporadic ALS is the most prevalent form of ALS consisting up to 90 to 95 percent of all ALS cases. [11] This type of ALS occurs sporadically with no known family history. Although the etiology of sporadic ALS is unknown, epidemiological data indicate that environmental & genetic factors contribute to its pathogenesis. Some of the risk factors that have increased ALS incidence are listed:
Several other factors such as pesticides, viruses, occupational workers (electrical workers, construction workers), dietary habits are proposed to be associated with ALS.
The etiology of sporadic ALS is complex. A combination of oxidative stress, mitochondrial dysfunction, glutamate excitotoxicity, inflammation, and apoptosis has been suggested as possible causes. Single-nucleotide polymorphisms (SNPs) in the paraoxonase gene cluster (PON) have been associated with sporadic ALS. PON enzymes are seen in insecticides, nerve gas agents, and in statin drugs. [18] Another example would be C9orF72 (Chromosome 9 open reading frame 72) which is a frequent cause of ALS, is responsible for about 10% of sporadic cases. [19] C9orf72 gene is also associated with familial ALS, Frontotemporal Dementia (FTD), and ALS with FTD. Few other examples of FALS genes mutations and variants occurring in sporadic ALS are ATXN2 gene, SETX gene, FUS gene. [19] These reports reinforce the concept that familial and sporadic ALS are not mutually exclusive categories but connected to each other.
Always! We start from scratch—no copying, no AI—just pure, human-written work with solid research and citations. You can even get a plagiarism report to confirm it’s 95%+ unique, ready for worry-free submission.
Learning in depth about the molecular mechanisms for the degeneration of motor neurons in ALS can help in better understanding of the disease and can provide insight into developing newer strategies and treatments. This section provides a brief overview of the molecular and cellular mechanisms that have been proposed to contribute to ALS pathogenesis.
6.1 Glutamate Excitotoxicity
Glutamate is an important excitatory neurotransmitter in the central nervous system. During a normal neurotransmission process, glutamate is released into the synaptic cleft, where it activates postsynaptic receptors. This activity is regulated by transporter proteins, called excitatory amino acid transporters (EAATs). [20] Excessive activation of glutamate receptors and failure in clearing the neurotransmitter from the synaptic cleft can induce injury to neurons. This abnormal receptor activity leads to a massive influx of calcium that triggers apoptotic pathways causing motor neuron death and degeneration.
6.2 Mitochondrial Dysfunction
You bet! From APA to IEEE, our writers nail every style with precision. Give us your guidelines, and we’ll craft a paper that fits your academic standards perfectly, no sweat.
Mitochondria are the most important organelles for cellular respiration, energy production, calcium homeostasis, and apoptosis. They represent a primary site for intracellular production of reactive oxygen species (ROS), a major source of oxidative stress, that impairs the normal functioning of mitochondria. Hence, any structural alterations or mutations in mitochondria can lead to the pathogenesis of ALS. [21]
6.3 Oxidative Stress
Free radicals or ROS are natural byproducts of oxygen metabolism. Oxidative stress is caused when the production of ROS is greater than the capacity of cells to remove them. This excessive accumulation of ROS causes permanent damage to cell structures, DNA, and RNA causing motor neuron degeneration. SOD1 is an important protein to prevent oxidative damage in cells. [21] Mutation in the SOD1 gene causes disruption of cellular functions and cytotoxicity. This can be confirmed by testing the levels of oxidative stress. 3-nitrotyrosine (3-NT) is an established biomarker for oxidative stress and the levels of 3-NT is elevated in serum, urine, and CSF samples of ALS patients. [22]
Yep! Use our chat feature to tweak instructions or add details anytime—even after your writer’s started. They’ll adjust on the fly to keep your essay on point.
6.4 Neuroinflammation
A familiar characteristic of ALS and other neurodegenerative diseases is the neuroinflammation. In the CNS, microglial cells are macrophages that act as the first line of defense against infections or injuries, thereby protecting motor neurons and astrocytes. Microglial cells have immunological properties, that can be either beneficial or harmful to motor neuron survival. As ALS progresses and the motor neuron damage worsens, the astrocytes and motor neurons release mutated SOD1 proteins that stimulate the activation of microglial cells. Activated microglial cells cause switch from neuroprotective and anti-inflammatory to a neurotoxic and pro-inflammatory phenotype. [21], [22]
The early and accurate diagnosis of ALS can be challenging because of the complex and heterogeneous nature of ALS. There are no definitive diagnostic tests to prove ALS, hence differential diagnosis and investigations are conducted for an individual patient. This includes obtaining a thorough patient history, conducting neuroimaging scans, electromyography, laboratory tests, and genetic testing.
Easy—place your order online, and your writer dives in. Check drafts or updates as you go, then download the final paper from your account. Pay only when you’re happy—simple and affordable!
In the late 1990s, diagnostic criteria were developed to standardize diagnosis of ALS and research studies for clinical trials.
Grade 1: Able to work or perform housework;
Grade 2: Independent living but unable to work;
Grade 3: Requiring assistance for eating, excretion, or ambulation;
Super fast! Our writers can deliver a quality essay in 24 hours if you’re in a pinch. Pick your deadline—standard is 10 days, but we’ll hustle for rush jobs without skimping.
Grade 4: Presence of respiratory insufficiency, difficulty in coughing out sputum, or dysphagia;
Grade 5: Using a tracheostomy tube, tube feeding, or tracheostomy positive-pressure ventilation.
The primary diagnosis of ALS is conducted by clinical examination and series of diagnostic tests that help to exclude diseases that mimic ALS. Some of the conditions that mimic ALS are Cervical spondylotic myelopathy, Kennedy disease (KD), Multiple Sclerosis, Parkinson’s diseases, and Post-polio syndrome (PPS). The following table shows a list of differential diagnosis and clinical overlap with ALS; [24]
| Differential diagnosis of ALS | Clinical overlap with ALS | Diagnostic test to rule out |
| Kennedy syndrome | Progressive motor neuron degeneration | Genetic testing, blood test for identification of specific mutations |
| Huntington disease | Progressive motor disturbances and involuntary movements | Genetic testing |
| Post-polio progressive muscular atrophy | Double vision, droopy eyelids, muscle weakness | EMG, blood tests |
| Parkinson’s disease | Progressive motor dysfunction and bradykinesia | Combination of laboratory tests and neuroimaging |
| Multiple sclerosis | Sensory loss and muscle weakness | Neuroimaging and spinal tap |
For example, Kennedy disease (KD), known as spinobulbar muscular atrophy, is an X-linked disorder of brainstem and spinal cord. KD symptoms demonstrate slow progressive LMN signs in the bulbar region and proximal limbs, fasciculations, mild cognitive impairment, sensory disturbance, and gynecomastia. [24] In addition, increased creatine kinase (CK) levels and low amplitude of sensory nerve action can help to differentiate KD from ALS. Progression of KD is slower than that of typical ALS. To confirm this diagnosis, a genetic test for detection for KD is also required.
Definitely! From astrophysics to literary theory, our advanced-degree writers thrive on tough topics. They’ll research deeply and deliver a clear, sharp paper that meets your level—high school to Ph.D.
Although the essential diagnostic criteria of ALS are defined by the El Escorial criteria, many misdiagnoses still occur. ALS mimic syndromes can be in terms of the anatomy, symptoms, or clinical manifestations. There is no single confirmatory test for ALS, but an extensive workup can help to rule out differential diagnosis. A comprehensive diagnostic workup includes most of the following procedures: [23]
A definitive diagnosis of ALS requires evidence of LMN and UMN degeneration, signs of progression, and spread of neurological symptoms within the anatomical region. The electrophysiological, laboratory, and neuroimaging results should not show evidence of any other pathological symptoms that mimic ALS.
Muscle strength and function are considered the most important endpoints that show effectiveness and consistency for any symptomatic treatment. For ALS, they can be assessed by;
Other scales that measure functional disability are the Norris scale, the Appel Scale, and the Pinch grip strength method. However, the ALSFRS-R is the preferred scale. If it is not used as primary endpoint, it can be used as a secondary one.
Although pathological mechanisms have been explained, ALS remains incurable disease because of failure of clinical trials and lack of any effective therapy. The rapid advancement in genetic discoveries in ALS emphasizes the point that ALS is a multi-subtype syndrome rather than a single disease. This can be one of the reasons why many previous clinical trials have failed. This section will review the recent developments in therapeutic compounds and alternative therapies for ALS.
We tailor your paper to your rubric—structure, tone, everything. Our writers decode academic expectations, and editors polish it to perfection, ensuring it’s grade-ready.
As the pathogenesis of ALS is complex, there is no effective treatment to cure ALS. Nevertheless, there are several therapeutic strategies can slow the progression of symptoms, prevent complications, and prolong survival.
The FDA approved Radicava in May 2017 in the United States, based on a six-month clinical trial conducted in Japan and granted it an Orphan drug designation. Radicava was discovered and developed by Mitsubishi Tanabe Pharma Corporation and will be commercialized in the United States by MT Pharma America. [26] It has been approved as a treatment option for ALS in Japan and South Korea. The drug is primarily known to slow the decline in physical function and delay the progression of ALS.
Upload your draft, tell us your goals, and our editors will refine it—boosting arguments, fixing errors, and keeping your voice. You’ll get a polished paper that’s ready to shine.
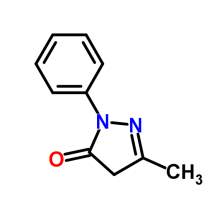 Edaravone is an active ingredient in Radicava, which is a member of the substituted 2-pyrazolin-5-one class. The chemical name of edaravone is 3-methyl-1-phenyl-2-pyrazolin-5-one. The molecular formula is C10H10N20 and the molecular weight is 174.20. Edaravone is a white crystalline powder and has a melting point of 129.7°C. It is soluble in acetic acid, ethanol, methanol, and slightly soluble in water. [27] Radicava injection supplied for intravenous infusion contains 30 mg edaravone in 100 mL isotonic, sterile, aqueous solution.
Edaravone is an active ingredient in Radicava, which is a member of the substituted 2-pyrazolin-5-one class. The chemical name of edaravone is 3-methyl-1-phenyl-2-pyrazolin-5-one. The molecular formula is C10H10N20 and the molecular weight is 174.20. Edaravone is a white crystalline powder and has a melting point of 129.7°C. It is soluble in acetic acid, ethanol, methanol, and slightly soluble in water. [27] Radicava injection supplied for intravenous infusion contains 30 mg edaravone in 100 mL isotonic, sterile, aqueous solution.
Radicava (Edaravone/MCI-186) is a neuroprotective drug that has properties of a free radical scavenger. Since oxidative stress is one of the major cause ALS, removal of free radicals may offer therapeutic benefits. Several free radical scavengers have been assessed for their efficacy, but only a few drugs have shown success in studies conducted. Edaravone is known to eliminate lipid peroxides and hydroxyl radicals and protects neurons from increased oxidative stress. [28] A reduced concentration of an oxidative stress biomarker, 3-nitrotyrosine, is seen in the cerebrospinal fluid. Edaravone can readily cross the blood-brain barrier (BBB), thereby explaining its efficacy while other scavengers have failed to demonstrate such effectiveness. [29] Thus, treatment with Edaravone slows the progression of functional motor disturbances in ALS patients
Sure! Need ideas? We’ll pitch topics based on your subject and interests—catchy and doable. Pick one, and we’ll run with it, or tweak it together.
The toxicity potential of edaravone has not been assessed adequately. There is no reported evidence of carcinogenic abnormalities. Edaravone was also tested negative for bacterial reverse mutation in Chinese hamster lung chromosomal aberration (in vitro) and mouse micronucleus assays (in vivo). Intravenous administration of edaravone had no effect on fertility. However, disruption of the estrus cycle and irregular mating behavior was noted at the highest dose tested.
Some common adverse reactions that occurred in Edaravone treated patients were contusion, gait disturbance, and headaches. Adverse effects of edaravone were observed in embryo development studies that include decrease in fetal body weight, delays in markers of development, and increased mortality. There were a few deaths in edaravone controlled studies as well as in placebo-treated patients. These deaths were related to respiratory failure which is the most common cause of death in ALS.
Population PK analysis indicates that the pharmacokinetics of edaravone is not affected by gender, age, race, or weight. There is a fetal developmental risk associated with the use of edaravone in pregnant women. However, there is no adequate data to prove the presence of edaravone in human milk, effects of the drug on milk production, or on the breastfed infant. No safety concerns related to hepatic or renal impairment has been reported.
The edaravone clinical development program for edaravone started in 2001for treatment of ALS in Japan. The ALS clinical trial program was conducted to explore efficacy and safety of edaravone and consisted of one Phase II and four Phase III studies. All the clinical trials were conducted in accordance with Good Clinical Practice and the guiding principles of the Declaration of Helsinki.
Yes! If you need quick edits, our team can turn it around fast—hours, not days—tightening up your paper for last-minute perfection.
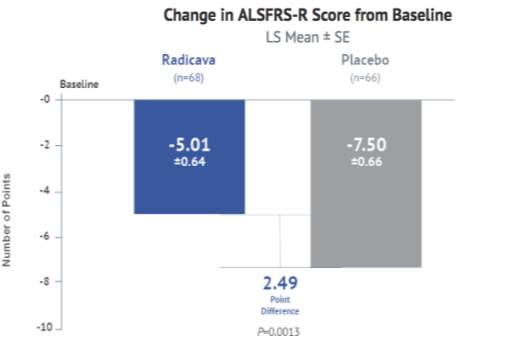
The following table provides a snapshot of the important clinical trial information for Edaravone.
Table 1: Studies in the ALS Development Program
| Study No. | Study Description | Study Design | No of Patients | Dosage Period |
| MCI186-12 | A Phase II exploratory study of edaravone in subjects with ALS | Open-labeled uncontrolled | 19 subjects |
You Want The Best Grades and That’s What We Deliver
Our top essay writers are handpicked for their degree qualification, talent and freelance know-how. Each one brings deep expertise in their chosen subjects and a solid track record in academic writing.
We offer the lowest possible pricing for each research paper while still providing the best writers;no compromise on quality. Our costs are fair and reasonable to college students compared to other custom writing services.
You’ll never get a paper from us with plagiarism or that robotic AI feel. We carefully research, write, cite and check every final draft before sending it your way.